Protein molecules carry out a vast array of biological functions. Indeed, almost every cellular process, from genome regulation to energy metabolism, requires a unique set of proteins with their precise concentration in a cell. Understanding the details of the process of protein synthesis and its regulation remains one of the fundamental questions of genetics and evolutionary cell biology. In our lab, we study this by integrating the theoretical and computational models of protein synthesis with high-throughput sequencing data.

Recent Publications:
Dual influence of transcriptional and translational kinetics on gene expression noise
Gene expression noise, the inherent randomness in protein levels, plays a crucial role in cellular heterogeneity and diverse responses to environmental conditions. While transcriptional bursts and their impact on noise have been extensively studied over the past two decades, recent experiments suggest that mRNA folding-unfolding kinetics can also create bursts in gene translation. This means mRNA can switch between active and inactive states, while gene translation occurs only when mRNA is in the active state. However, the combined influence of transcriptional and translational bursts on gene expression noise remains largely unexplored. To address this, we present a gene expression model that integrates both transcriptional and translational bursting phenomena to provide a detailed understanding of the factors governing protein noise. Using the generating function technique, we solve the model and derive analytical expressions for mean protein levels and noise in protein copy numbers. Our findings show that translational bursting significantly contributes to overall noise and the noise from transcriptional bursts further amplifies the relative contribution of translational noise. We also find that translation efficiency, defined as the rate of protein synthesis from a single mRNA transcript, increases the overall noise of gene expression at fixed protein levels. This dependence of gene expression noise on translation efficiency suggests that both translation and transcription kinetics are required to be tuned simultaneously to achieve specific protein levels and gene expression noise. Thus, this study improves our understanding of the factors regulating gene expression noise, providing insights into how transcriptional and translational bursting shape this noise.
Ref. Phys. Rev. E 112, 044410 (2025)
Speed-Energy-Efficiency Trade-off in Hsp70 Chaperone System
Proteins must fold into their native structure to carry out cellular functions. However, they can sometimes misfold into non-native structures, leading to reduced efficiency or malfunction. Chaperones help prevent misfolding by guiding proteins to their active state using energy from ATP hydrolysis. Experiments have revealed numerous kinetic and structural aspects of how various chaperones facilitate the folding of proteins into their native structure. However, what remains missing is a fundamental theoretical understanding of their operational mechanisms, especially the limits and constraints imposed on their efficiency by energy flow and dissipation. To address this, we built a kinetic model of the Hsp70 chaperone system by incorporating all key structural and kinetic details. Then, using the chemical kinetic equations, we investigate how energy expenditure shapes the efficiency of Hsp70 chaperones in the proper folding of misfolded proteins. We show that ATP consumption by chaperones significantly enhances the folding of proteins into their native states. Our investigations reveal that a chaperone achieves optimal efficiency when its binding to misfolded proteins is much faster than the misfolding kinetics of that protein. We also demonstrate the presence of an upper bound on a chaperone’s efficiency of protein folding and its overall rescue rate. This upper bound increases with energy dissipation until it reaches a saturation point. Furthermore, we show a speed-energy-efficiency trade-off in chaperone action, demonstrating that it is impossible to simultaneously optimize the efficiency of chaperone-assisted protein folding and the energy efficiency of the process.
Ref. J. Phys. Chem. B 2024, 128, 49, 12101–12113
Understanding the regulation of protein synthesis under stress conditions
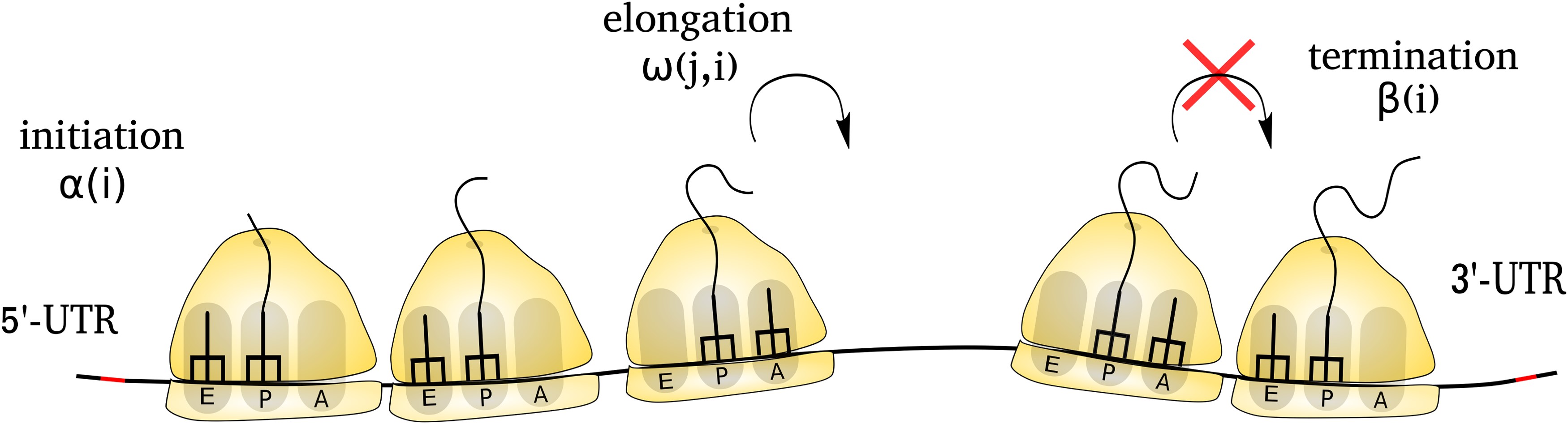
Protein synthesis regulation primarily occurs at translation initiation, the first step of gene translation. However, the regulation of translation initiation under various conditions is not fully understood. Specifically, the reason why protein production from certain mRNAs remains resistant to stress while others do not show such resilience. Moreover, why is protein production enhanced from a few transcripts under stress conditions, whereas it is decreased in the majority of transcripts? We address them by developing a Monte Carlo simulation model of protein synthesis and ribosome scanning. We find that mRNAs with strong Kozak contexts exhibit minimal reduction in translation initiation rate under stress conditions. Moreover, these transcripts exhibit even greater resilience to stress when the scanning speed of 43S ribosome subunit is slow, albeit at the cost of reduced initiation rate. This implies a trade-off between initiation rate and the ability of mRNA to withstand stress. We also show that mRNAs featuring an upstream ORF can act as a regulatory switch. This switch elevates protein production from the main ORF under stress conditions; however, minimal to no proteins are produced under the normal condition. Because, in stress, a larger fraction of 43S ribosomes bypasses the upstream ORF due to its weak Kozak context. This, in turn, increases the number of scanning ribosomes reaching the main ORF, whose strong Kozak context can convert them into 80S ribosomes, even under stress conditions. This switching allows an efficient use of cellular resources by producing proteins when they are required. Thus, our computational study provides valuable insights into our understanding of stress-responsive translation-initiation.
Ref. Biophysical Journal, Volume 123, Issue 20, 3627 – 3639 (2024)
We appreciate receiving financial support from the following funding agencies listed below.

