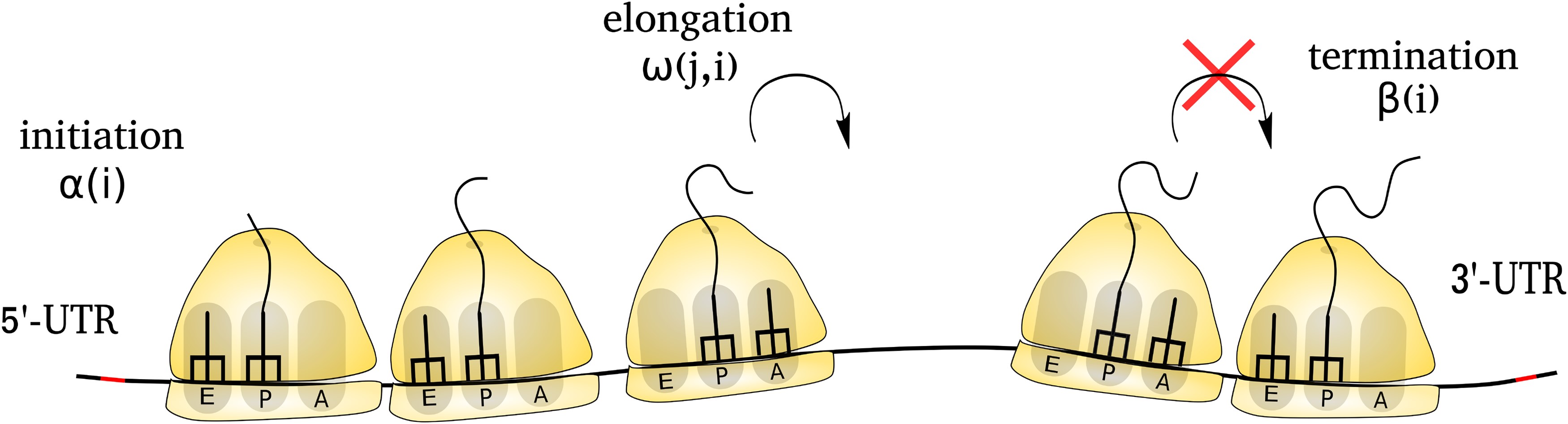
Protein molecules carry out a vast array of biological functions. Indeed, almost every cellular process, from genome regulation to energy metabolism, requires a unique set of proteins with their precise concentration in a cell. Understanding the details of the process of protein synthesis and its regulation remains one of the fundamental questions of genetics and evolutionary cell biology. In our lab, we study this by integrating the theoretical and computational models of protein synthesis with high-throughput sequencing data.

Recent Publications:
Decoding stoichiometric protein synthesis in E. coli through translation rate parameters
E. coli is one of the most widely used organisms for understanding the principles of cellular and molecular genetics. However, we are yet to understand the origin of several experimental observations related to the regulation of gene expression in E. coli. One of the prominent examples in this context is the proportional synthesis in multiprotein complexes where all of their obligate subunits are produced in proportion to their stoichiometry. In this work, by combining the next-generation sequencing data with the stochastic simulations of protein synthesis, we explain the origin of proportional protein synthesis in multicomponent complexes. We find that the estimated initiation rates for the translation of all subunits in those complexes are proportional to their stoichiometry. This constraint on protein synthesis kinetics enforces proportional protein synthesis without requiring any feedback mechanism. We also find that the translation initiation rates in E. coli are influenced by the coding sequence length and the enrichment of A and C nucleotides near the start codon. Thus, this study rationalizes the role of conserved and nonrandom features of genes in regulating the translation kinetics and unravels a key principle of the regulation of protein synthesis.
Ref. Biophysical Reports 3.4 (2023)
Optimization of ribosome utilization in Saccharomyces cerevisiae
Resource optimization in protein synthesis is often looked at from the perspective of translation efficiency—the rate at which proteins are synthesized from a single transcript. The higher the rate of protein synthesis, the more efficiently a transcript is translated. However, the production of a ribosome consumes significantly more cellular resources than an mRNA molecule. Therefore, there should be a stronger selection pressure for optimizing ribosome usage than translation efficiency. This paper reports strong evidence of such optimization which becomes more prominent in highly expressed transcripts that consume a significant amount of cellular resources. The ribosome usage is optimized by the biases in codon usage and translation initiation rates. This optimization significantly reduces the ribosome requirement in Saccharomyces cerevisiae. We also find that a low ribosome density on mRNA transcripts helps optimize ribosome utilization. Therefore, protein synthesis occurs in a low ribosome density regime where translation–initiation is the rate-limiting step. Our results suggest that optimizing ribosome usage is one of the major forces shaping evolutionary selection pressure, and thus provide a new perspective to resource optimization in protein synthesis.
Ref: PNAS Nexus 2, pgad074 (2023)
The presence of a single cluster of nonoptimal codons was found to decrease a transcript’s half-life through the interaction of the ribosome-associated quality control machinery with stalled ribosomes in Saccharomyces cerevisiae. The impact of multiple nonoptimal codon clusters on a transcript’s half-life, however, is unknown. Using a kinetic model, we predict that inserting a second nonoptimal cluster near the 5′ end can lead to synergistic effects that increase a messenger RNA’s (mRNA’s) half-life in S. cerevisiae. Specifically, the 5′ end cluster suppresses the formation of ribosome queues, reducing the interaction of ribosome-associated quality control factors with stalled ribosomes. We experimentally validate this prediction by introducing two nonoptimal clusters into three different genes and find that their mRNA half-life increases up to fourfold. The model also predicts that in the presence of two clusters, the cluster closest to the 5′ end is the primary determinant of mRNA half-life. These results suggest the “translational ramp,” in which nonoptimal codons are located near the start codon and increase translational efficiency, may have the additional biological benefit of allowing downstream slow-codon clusters to be present without decreasing mRNA half-life. These results indicate that codon usage bias plays a more nuanced role in controlling cellular protein levels than previously thought.
Ref. PNAS 118, e2026362118 (2021)
We appreciate receiving financial support from the following funding agencies listed below.

